
8 Minuten
Etwas Winziges unterbrach einen tödlichen Handschlag innerhalb von Gehirnzellen. Die Konsequenz: Mitochondrien — die Kraftwerke der Zelle — begannen zu versagen, und Neuronen, die zuvor vor Energie strotzten, fingen an zu schwächeln. Dieses Zusammenspiel steht im Mittelpunkt einer neuen Studie der Case Western Reserve University School of Medicine, die einen direkten Weg von Alpha‑Synuclein‑Clustern zum mitochondrialen Zusammenbruch nachzeichnet und einen molekularen Köder vorstellt, um den Schaden zu stoppen.
Alpha‑Synuclein gilt seit Jahrzehnten als Verdächtiger bei der Parkinson‑Krankheit. Faltet sich dieses Protein fehl oder verklumpt es, leiden Neurone. Parallel beobachten Kliniker und Labors, dass Mitochondrien in betroffenen Neuronen geschwächt, energiearm und anfällig für das Auslösen des programmierten Zelltods sind. Wie aber diese beiden Phänomene zusammenhängen, war bislang unklar. Die neue Arbeit beleuchtet eine präzise biochemische Interaktion: Alpha‑Synuclein haftet an einem Enzym tief im Inneren der Mitochondrien namens ClpP, und diese Liaison scheint die Fähigkeit des Organells zu sabotieren, beschädigte Proteine zu entsorgen und das metabolische Gleichgewicht zu erhalten.
Was das Team herausfand und wie es getestet wurde
In Gewebeproben aus dem Labor und in Tiermodellen beobachteten die Forschenden, wie Alpha‑Synuclein an mitochondriales ClpP bindet und anschließend eine Störung der mitochondrialen Protein‑Qualitätskontrolle eintritt. Man kann sich ClpP als die Müllentsorgungsanlage des Kraftwerks vorstellen: Wird sie blockiert, sammeln sich Abfallprodukte an und der Motor stottert. Die nachgelagerten Effekte sind für Parkinson‑Forschende vertraut: Neurone verlieren ihre Effizienz, dopaminproduzierende Schaltkreise schwächen sich, Entzündungsprozesse nehmen zu, und Bewegungs‑ sowie kognitive Funktionen verschlechtern sich.
Statt Alpha‑Synuclein flächendeckend zu bekämpfen, entwarfen die Forschenden ein kurzes Peptid namens CS2, das als Köder fungiert. Die Idee ist einfach und elegant: Das fehlerhafte Protein vom ClpP weglocken, damit die Mitochondrien wieder ihrer Aufgabe nachgehen können. In ex vivo‑menschlichem Hirngewebe, in kultivierten Neuronen und in Mausmodellen reduzierte CS2 Entzündungsmarker, bewahrte die mitochondriale Aktivität und führte zu messbaren Verbesserungen in motorischen und kognitiven Tests der Versuchstiere.
Methodisch stützte sich die Studie auf mehrere etablierte experimentelle Ansätze, um die Interaktion und funktionelle Folgen zu belegen:
- Biochemische Nachweise wie Co‑Immunpräzipitation und Protein‑Interaktionsassays, um direkte Bindungen zwischen Alpha‑Synuclein und ClpP zu demonstrieren.
- Proteomische Analysen und Western‑Blots, um Akkumulation beschädigter mitochondrialer Substrate und veränderte ClpP‑Aktivität festzuhalten.
- Funktionelle Messungen der mitochondrialen Atmung (z. B. Respirometrie, Seahorse‑Assays) zur Bestimmung von ATP‑Produktion, respiratorischer Kapazität und protonenabhängigen Prozessen.
- Zellbiologische Versuche in kultivierten Neuronen, einschließlich Lebend‑/Tot‑Färbungen, ROS‑Messungen (reaktive Sauerstoffspezies) und Markern für proteotoxischen Stress.
- Verhaltens‑ und kognitive Tests in Mausmodellen (z. B. Rotarod, Open‑Field, Gedächtnistests), um physiologische Konsequenzen einer CS2‑Intervention zu bewerten.
Die Ergebnisse zeigten konsistent, dass die Präsenz von aggregiertem Alpha‑Synuclein die ClpP‑Funktion hemmt, was zu einer Akkumulation defekter oder fehlgefalteter mitochondrialer Proteine führt. CS2 fungierte in experimentellen Settings als kompetitiver Interaktionspartner, der Alpha‑Synuclein bindet und so die Blockade von ClpP verhindert. Dadurch normalisieren sich Parameter der mitochondrialen Homöostase und die Zellen zeigen weniger Zeichen von Entzündungsreaktionen und oxidativem Stress.
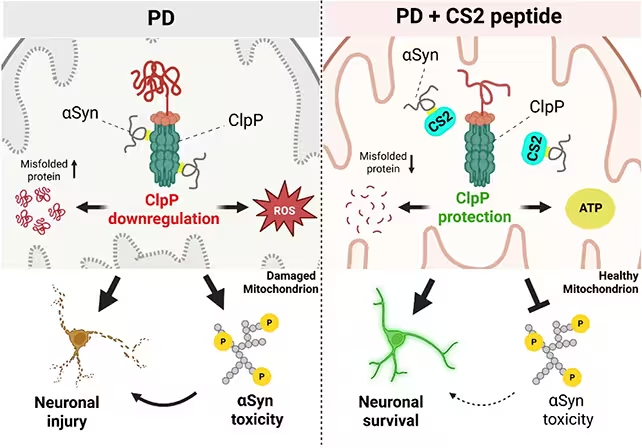
CS2 konnte das ClpP‑Enzym schützen.
Dieser protektive Effekt ist bedeutsam, weil er einen mechanistischen Schritt adressiert, der vermutlich stromaufwärts vieler der zellulären Ausfälle bei Parkinson liegt. Wenn die Interaktion von Alpha‑Synuclein mit ClpP einer der frühen Fehltritte in der Krankheitskaskade ist, könnte das Abschirmen von ClpP mehrere nachgelagerte Schäden stoppen oder verlangsamen.
Wissenschaftlicher Kontext und Implikationen
Parkinson ist keine Einbahnstraße mit nur einem Auslöser. Gene, Umweltfaktoren, Alterungsprozesse und zelluläre Stressfaktoren verknüpfen sich auf komplexe Weise. Die neue Studie erhebt keinen Anspruch auf ein universelles Heilmittel. Sie liefert jedoch Klarheit: einen definierten molekularen Kontaktpunkt, der verändert werden kann. Eine Intervention an dieser Stelle könnte komplementär zu Strategien wirken, die Alpha‑Synuclein‑Aggregation verringern, mitochondrialen Biogenese‑Programme stärken oder Entzündungsreaktionen modulieren.
Konkrete Implikationen für Forschung und mögliche klinische Entwicklung umfassen mehrere Bereiche:
- Therapeutische Targeting‑Strategien: CS2 ist ein Prototyp für sogenannte Interaktionsblocker. Solche Moleküle können weiter optimiert werden — etwa hinsichtlich Stabilität, Zellpermeabilität und Target‑Spezifität.
- Lieferung in das Gehirn: Ein großes praktisches Hindernis ist die Zugänglichkeit empfindlicher Neurone. Peptide wie CS2 müssen entweder so modifiziert werden, dass sie die Blut‑Hirn‑Schranke überqueren, oder es sind geeignete Trägersysteme (z. B. Nanopartikel, virale Vektoren mit Peptidkapseln) zu entwickeln.
- Sicherheitsprofil und Off‑Target‑Effekte: Jede Substanz, die Protein‑Protein‑Interaktionen innerhalb von Mitochondrien verändert, muss sorgfältig auf unerwünschte Wirkungen geprüft werden. Kleine Veränderungen in der Proteostase können weitreichende Folgen haben.
- Kombinationstherapien: Das Abschirmen von ClpP könnte in Kombination mit Therapien sinnvoll sein, die Aggregation reduzieren, mitochondriale Biogenese fördern (z. B. durch PGC‑1α‑Aktivierung) oder Neuroinflammation dämpfen.
Das Forschungsteam schätzt eine Vorlaufzeit von etwa fünf Jahren, bevor CS2 oder ähnliche Biologika in erste Studien am Menschen gelangen könnten. Dieser Zeitrahmen ist für eine auf Peptiden basierende Therapie nicht unrealistisch, doch geboten ist Vorsicht: präklinische Langzeitsicherheit, pharmakokinetische Optimierung und umfassende toxikologische Tests sind erforderlich, bevor klinische Prüfungen starten können.
Ein weiteres wichtiges Diskussionsthema ist die Heterogenität der Parkinson‑Pathologie: Nicht alle Patienten zeigen identische molekulare Muster. Daher ist es denkbar, dass ein ClpP‑zentrierter Ansatz besonders für Subgruppen sinnvoll ist — etwa Patientinnen und Patienten mit starker mitochondrieller Dysfunktion oder spezifischen Alpha‑Synuclein‑Pathologien.
Technische Feinheiten und Validierungsbedürfnisse
Um das Konzept weiter zu untermauern, sind mehrere technische Schritte empfehlenswert:
- Reproduktionsstudien in verschiedenen Tiermodellen, einschließlich transgener Modelle mit menschlichem Alpha‑Synuclein sowie sporadischen Modellen.
- Langzeitstudien zur Beurteilung der Wirkungsdauer von CS2, möglicher Immunantworten auf das Peptid und potenzieller Toxizitäten in peripheren Organen.
- Untersuchungen zu Interaktionen zwischen ClpP und anderen mitochondrialen Protease‑Systemen, um Kompensationsmechanismen zu verstehen.
- Analysen von Biomarkern im Blut oder Liquor, die frühzeitig Hinweise auf mitochondriale Verbesserung geben könnten (z. B. metabolische Marker, inflammatorische Zytokine, mitochondriale DNA‑Freisetzung).
Insgesamt verschiebt die Studie die tektonischen Platten der Parkinson‑Forschung zwar nicht dramatisch, aber sie legt ein neues, konkret angreifbares Ziel offen. Für Betroffene und ihre Familien bedeutet schon diese inkrementelle Klarheit etwas: Therapien, die den neuronalen Energiestoffwechsel erhalten, könnten zu längeren, funktional besseren Lebensphasen führen.
Fachliche Einschätzung und nächste Schritte
„Diese Arbeit ist eine seltene Verbindung von mechanistischer Präzision und therapeutischer Phantasie“, sagt Dr. Elaine Ramirez, eine fiktive Neurobiologin, die mitochondriale Dynamik untersucht. „Einen Köder zu entwerfen, um ein pathogenes Protein an der mitochondrialen Tür abzufangen, ist klug. Die Herausforderung besteht nun in der Verabreichung — genug Peptid in verletzliche Neurone zu bringen, ohne andere Qualitätskontrollsysteme der Zelle zu schwächen.“
Die Publikation erschien in Molecular Neurodegeneration und öffnet mehrere Entwicklungsachsen: die Optimierung der Stabilität von CS2, langfristige Sicherheitsstudien und die Erforschung, ob ähnliche Köder auch andere mitochondriale Enzyme schützen können. Sie wirft außerdem eine größere Frage auf: Wenn wir Mitochondrien davor bewahren, ihre Energie zu verlieren, könnten wir dann die Parkinson‑Trajektorie von einem unaufhaltsamen Fortschreiten zu einem behandelbaren oder zumindest verlangsamten Zustand verändern? Die kommenden Experimente werden entscheidend sein; vorläufig bietet das Feld jedoch einen frischen Ansatzpunkt.
Herausforderungen für Translation und Klinik
Bei der Translation in die Klinik stehen mehrere Hürden im Vordergrund:
- Pharmakokinetik und Verabreichung: Peptide werden im Körper oft schnell abgebaut. Modifikationen wie PEGylierung, Cyclisierung oder die Nutzung von Trägern könnten die Halbwertszeit verbessern.
- Blut‑Hirn‑Schranke: Strategien zur effizienten Überwindung der Blut‑Hirn‑Schranke sind zentral — sei es durch fokussierte Ultraschalltechniken, BBB‑modifizierende Liganden oder durch invasive Delivery‑Methoden bei schweren Fällen.
- Spezifität: Sicherstellen, dass CS2 nicht unbeabsichtigt andere mitochondriale Proteine bindet, die für die Zellgesundheit essenziell sind.
- Patientenselektion: Identifikation klinischer oder molekularer Biomarker, die jene Patientengruppen markieren, welche am ehesten von einem ClpP‑schützenden Ansatz profitieren.
Solche Herausforderungen sind keineswegs neu in der neurodegenerativen Forschung, doch die klare Mechanik hinter dem CS2‑Konzept macht zielgerichtete Optimierungen möglich und gibt Forschenden konkrete Messgrößen an die Hand.
Schlussbemerkung
Die Studie liefert nicht das endgültige Heilversprechen, aber sie ergänzt das Feld um eine präzise, testbare Hypothese: eine direkte Verbindung von Alpha‑Synuclein‑Aggregation zur mitochondrialen Dysfunktion über die Blockade von ClpP, und die Möglichkeit, diese Interaktion durch einen Peptid‑Köder wie CS2 zu unterbrechen. Als Baustein in einem multimodalen Therapieansatz — kombiniert mit Aggregationshemmern, Entzündungsmodulatoren und Strategien zur Stärkung der mitochondrialen Biogenese — könnte ein solcher Mechanismus dazu beitragen, Parkinson‑Verläufe zu verändern.
Für die wissenschaftliche Gemeinschaft heißt das: weiter experimentieren, robust replizieren und mit Bedacht in die translationalen Schritte gehen. Für Betroffene bleibt Hoffnung, aber keine Garantie; die nächsten Jahre werden zeigen, ob der molekulare Köder auf lange Sicht hält, was die präklinischen Ergebnisse versprechen.
Quelle: sciencealert
Kommentar hinterlassen