9 Minuten
Wissenschaftler haben eine einzelne Genvariante identifiziert, die eine außergewöhnlich seltene Form von Diabetes bei Neugeborenen verursacht – ein Befund, der neue Einblicke liefert, wie insulinproduzierende Beta-Zellen entstehen und versagen. Die Mutation schädigt Beta-Zellen und kann zu ihrem Absterben führen, was ein charakteristisches Dreiersyndrom bei betroffenen Säuglingen erklärt.
Ein Gen, ein verheerendes Kindheitssyndrom
Ein internationales Forscherteam konnte neonatalen Diabetes bei sechs Säuglingen auf Mutationen im Gen TMEM167A zurückführen. Diese Kinder zeigten zudem Mikrozephalie (eine ungewöhnlich kleine Kopfgröße) und in fünf der sechs Fälle Epilepsie – ein Merkmalbündel, das bereits als MEDS (Microcephaly, Epilepsy, and Diabetes Syndrome) bekannt ist. Bislang wurden nur zwei Gene (IER3IP1 und YIPF5) eindeutig mit MEDS in Verbindung gebracht. Die neuen Ergebnisse etablieren TMEM167A als dritte genetische Ursache.
Die Identifizierung eines weiteren MEDS-Verursachers erweitert das genetische Spektrum dieser seltenen Erkrankung und hilft, die molekularen Mechanismen zu entschlüsseln, die sowohl die neuronale Entwicklung als auch die Funktion der Bauchspeicheldrüse beeinflussen. Solche Erkenntnisse sind entscheidend, um klinische Diagnosen zu verfeinern, genetische Beratung zu ermöglichen und mögliche Therapieansätze zielgerichtet zu erforschen.
Wie TMEM167A die Insulinproduktion beeinträchtigt
TMEM167A wird sowohl in der Bauchspeicheldrüse als auch im Gehirn von Menschen und Mäusen exprimiert – eine Verteilung, die erklärt, warum Mutationen beide Organe betreffen können. Um die Auswirkungen der Mutation zu untersuchen, bearbeiteten die Forscher menschliche pluripotente Stammzellen: Das normale TMEM167A-Gen wurde durch die in einem MEDS-Patienten gefundene Variante ersetzt. Diese Stammzellen wurden anschließend differenziert, sodass sie sich in pankreatische Beta-Zellen entwickelten, also in die spezialisierten Zellen, die Insulin produzieren und sekretieren.
Die Verwendung humaner pluripotenter Stammzellen und gentechnischer Methoden wie gezielter Geneditierung (zum Beispiel CRISPR-basierte Ansätze) erlaubt es, krankheitsassoziierte Varianten im menschlichen Zellkontext zu modellieren. Solche Modelle sind besonders wertvoll, weil sie die direkten Folgen einer spezifischen Mutation auf Zellentwicklung, Zelldifferenzierung und Zellfunktion sichtbar machen, ohne auf interspezifische Unterschiede zwischen Tiermodellen angewiesen zu sein.
Entwicklung scheint normal – Funktion aber nicht
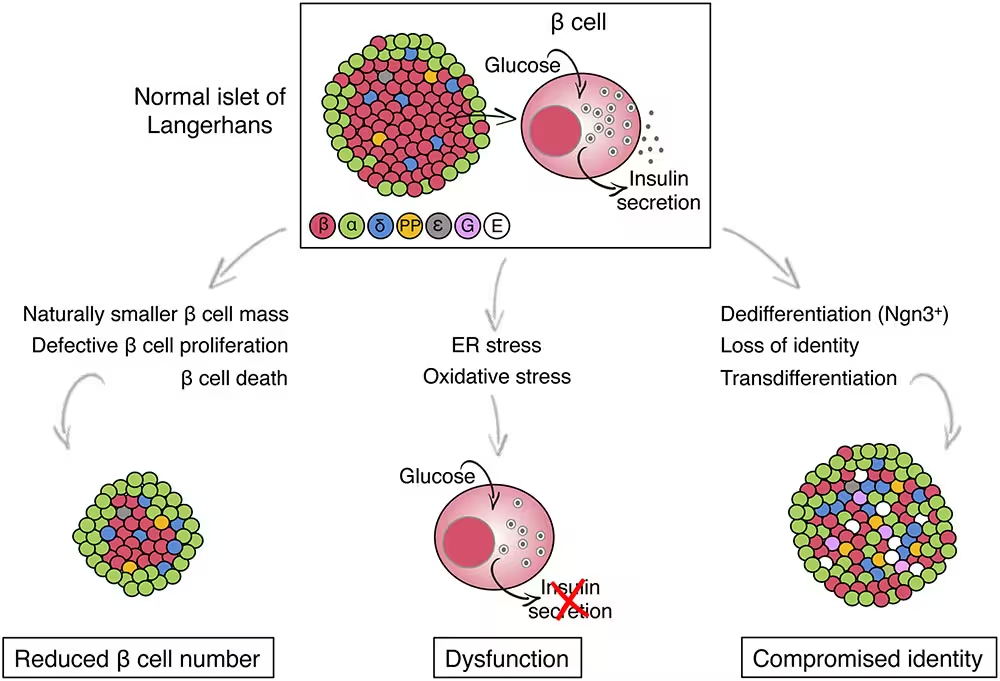
Überraschenderweise bildeten sich Beta-Zellen mit der TMEM167A-Variante zahlenmäßig und im Erscheinungsbild ähnlich wie gesunde Zellen, doch sie waren funktionell mangelhaft. Bei Glucose-Belastung setzten diese Zellen nicht wie gesunde Beta-Zellen Insulin frei. Die Fehlfunktion ließ sich auf Stress im endoplasmatischen Retikulum (ER) zurückführen – dem intrazellulären Netzwerk für Proteinfaltung und -transport. Anhaltender ER-Stress aktivierte Signalwege, die schließlich den Zelltod der Beta-Zellen auslösten.
Auf zellbiologischer Ebene sind anhaltender ER-Stress und die fehlgeschlagene Anpassungsreaktion (Unfolded Protein Response, UPR) bekannte Ursachen für den Funktionsverlust von Sekretionszellen. Bei Beta-Zellen kann eine fehlgeleitete UPR zur Aktivierung pro-apoptotischer Mediatoren wie CHOP führen und über längere Zeiträume den Verlust funktionsfähiger Zellen begünstigen.
„Die Fähigkeit, insulinproduzierende Zellen aus Stammzellen zu gewinnen, hat es uns ermöglicht zu untersuchen, was in den Beta-Zellen von Patienten mit seltenen sowie anderen Formen von Diabetes dysfunktional ist“, erläuterte die Diabetologin Miriam Cnop von der Freien Universität Brüssel. „Dies ist ein außergewöhnliches Modell, um Krankheitsmechanismen zu studieren und mögliche Therapien zu testen.“
Technisch gesehen erlaubte das Experimentieren mit humanen Stammzellmodellen die Beobachtung mehrerer relevanter Parameter: Genexpression während der Differenzierung, sekretorische Antwort auf Glukosestimulation, Marker für ER-Stress und Anzeichen für Apoptose. Die Kombination dieser Daten liefert ein umfassendes Bild davon, wie eine einzelne Genveränderung die komplexe Biologie von Beta-Zellen stören kann.
Warum das über seltene Erkrankungen hinaus wichtig ist
Obwohl MEDS extrem selten ist (vor dieser Studie waren nur etwa 11 Fälle dokumentiert), spiegeln die zellulären Probleme – ER-Stress, fehlende Insulinsekretion und Verlust von Beta-Zellen – Prozesse wider, die auch bei häufigeren Diabetesformen wie Typ-2-Diabetes beobachtet werden. Diese Überschneidungen bedeuten, dass Erkenntnisse über TMEM167A weitergehende Relevanz für die Diabetesforschung haben könnten, etwa bei der Identifizierung therapeutischer Ziele oder der Verbesserung von Stammzell-basierten Ersatztherapien für Beta-Zellen.
Besonders relevant ist der mechanistische Einblick: Wenn eine seltene monogene Mutation einen klaren Pathomechanismus (ER-Stress → gestörte Insulinsekretion → Beta-Zellverlust) liefert, kann dies als Modell dienen, um konservierte molekulare Schwachstellen zu identifizieren, die auch bei multifaktoriellen Erkrankungen eine Rolle spielen. Solche Gemeinsamkeiten lassen neue Hypothesen für die Pathogenese von Typ-2-Diabetes oder anderen Beta-Zell-Defekten entstehen.
„Die Identifikation der DNA-Veränderungen, die Diabetes bei Säuglingen auslösen, liefert uns einen einzigartigen Zugang, um Gene zu finden, die Schlüsselrollen in der Insulinproduktion und -sekretion spielen“, sagte die molekulargenetische Forscherin Elisa de Franco von der University of Exeter. „Unsere Arbeit zeigt, wie ein bislang wenig untersuchtes Gen wie TMEM167A entscheidend für die Insulinsekretion sein kann.“
Praktische Konsequenzen dieser Erkenntnisse reichen von verbesserten genetischen Screenings bei Neugeborenen mit Hypoglykämie oder atypischem Diabetesbild bis hin zur Suche nach kleinen Molekülen, die ER-Stress reduzieren oder die sekretorische Maschinerie stabilisieren. Auch die Übertragung von Erkenntnissen aus monogenen Syndromen auf polygenetische Erkrankungen ist ein bewährter Weg, um neue Therapieansätze zu entwickeln.
Ausblick: Diagnose und potenzielle Therapien
Klinisch ermöglicht die Erkennung von TMEM167A-Mutationen eine eindeutige genetische Diagnose von MEDS-Fällen, was für die Familienberatung, pränatale Analysen und die neonatale Versorgung von großer Bedeutung ist. Ein genetischer Befund kann behandelbare Komplikationen früher erkennbar machen und Familien eine bessere Grundlage für Entscheidungsfindung und Prognose geben.
Auf der Forschungsebene liefert das Stammzellmodell eine Testplattform für Wirkstoffe, die ER-Stress vermindern, die UPR modulieren oder die Insulinsekretion stärken könnten. Zu den möglichen Interventionsstrategien gehören pharmakologische Chaperone, kleine Moleküle, die die Proteinfaltung verbessern, sowie Substanzen, die spezifische UPR-Signalwege regulieren. Daneben sind genetische Korrekturen in patientenspezifischen Zellen (ex vivo) ein denkbarer, wenn auch langfristiger Ansatz für personalisierte Therapien.
Wichtig ist dabei die Einschätzung realistischer Zeithorizonte: Translational wirkende Therapien, die von Zellmodellen über präklinische Tests bis zur klinischen Anwendung gelangen, benötigen in der Regel mehrere Jahre Forschung, Sicherheitsprüfungen und klinische Studien. Dennoch öffnet die Studie einen direkten Weg von der Genentdeckung zur mechanistischen Einsicht und, perspektivisch, zu gezielten Interventionen.
Für die Zukunft sind mehrere Forschungsrichtungen naheliegend:
- Vertiefte Untersuchung der molekularen Rolle von TMEM167A in ER-Homeostase, Vesikelverkehr und Signaltransduktion.
- Screening existierender Wirkstoffbibliotheken auf Substanzen, die die sekretorische Funktion geschädigter Beta-Zellen verbessern.
- Entwicklung von Tiermodellen und humanen organoid-Systemen, um Gewebeübergreifende Effekte (Gehirn und Pankreas) besser zu verstehen.
- Ausbau genetischer Screenings für Neugeborene mit atypischen Symptomen, um die Prävalenz und das klinische Spektrum von MEDS besser abzuschätzen.
Solche Forschungswege können nicht nur Betroffenen mit MEDS helfen, sondern auch allgemeinere Fortschritte in der Diabetesforschung fördern – von Biomarkern bis zu Zelltherapien.
Technische und klinische Details zur Forschung
Das Experimentieren mit humanen pluripotenten Stammzellen erfordert präzise Protokolle zur Differenzierung in endokrine Pankreaszellen. Forscher überwachen während dieser Prozesse verschiedene Markerstadien: definitive Endoderm-Marker, Pankreas-Progenitor-Marker und schließlich Beta-Zell-spezifische Marker wie INS (Insulin), PDX1 und NKX6-1. Parallel werden funktionelle Tests durchgeführt, etwa die Glukose-stimulierbare Insulinsekretion (GSIS), um die tatsächliche sekretorische Kapazität zu prüfen.
ER-Stress-Indikatoren umfassen die Expression von Hitzeschockproteinen, chaperonähnlichen Molekülen und Transkriptionsfaktoren, die zur UPR gehören. Auf Signalwegebene sind PERK, IRE1 und ATF6 die Hauptachsen der UPR; ihre Aktivierung und das Gleichgewicht zwischen adaptiven und pro-apoptotischen Reaktionen bestimmen, ob eine Zelle den Stress übersteht oder in den programmierten Zelltod übergeht.
In vielen neuroendokrinen Erkrankungen zeigt sich außerdem eine Wechselwirkung zwischen gestörter Proteinverarbeitung, gestörtem Vesikelverkehr und erhöhtem oxidativem Stress. TMEM167A könnte an Kompartimentierung und Membranfluss beteiligt sein; seine Fehlfunktion könnte deshalb sowohl die Proteinfaltung als auch die Transportwege von Insulinvorläufern stören.
Limitierungen und offene Fragen
Trotz der starken Hinweise bleiben einige Fragen offen: Welche biochemische Funktion hat TMEM167A exakt auf molekularer Ebene? In welchem Ausmaß tragen Modifikatoren oder genetische Hintergrundvarianten zur Phänotypausprägung bei? Und wie übertragbar sind Befunde aus Zellkulturmodellen auf den Organismus und die klinische Realität?
Außerdem ist die Zahl dokumentierter Fälle sehr gering, sodass epidemiologische Aussagen zur Häufigkeit und Variabilität der Erkrankung vorerst begrenzt sind. Größere internationale Kooperationen und Register seltener Erkrankungen können helfen, diese Lücken zu schließen und die klinische Vielfalt von MEDS genauer zu beschreiben.
Nicht zuletzt sind Sicherheits- und Effektivitätsfragen bei möglichen Interventionen – sei es pharmakologisch oder genetisch – sorgfältig zu prüfen, insbesondere bei Patienten im Neugeborenenalter oder in sensiblen neurologischen Entwicklungsphasen.
Zusammenfassung und Bedeutung für die Diabetesforschung
Die Entdeckung der TMEM167A-Variante als Ursache eines MEDS-Falls liefert einen klaren Mechanismus, der von gestörter Proteinverarbeitung über ER-Stress zu mangelnder Insulinsekretion und Beta-Zellverlust führt. Diese Erkenntnis verbindet seltene monogene Syndrome mit allgemeinen pathophysiologischen Konzepten der Diabetesforschung und eröffnet neue Forschungs- und Diagnosewege.
Langfristig können solche genetischen Befunde dazu beitragen, individuellere Behandlungsansätze zu entwickeln, die sowohl seltene als auch häufigere Formen von Diabetes berücksichtigen. Gleichzeitig stärken sie das Vertrauen in Stammzell-basierte Modelle als zentrale Werkzeuge, um krankheitsrelevante Mechanismen zu untersuchen und potenzielle Therapeutika zu testen.
Die Studie ist ein Beispiel dafür, wie genetische Diagnostik, moderne Zellbiologie und translational orientierte Forschung Hand in Hand gehen, um sowohl die Grundlagenforschung als auch die klinische Versorgung seltener und häufiger Krankheiten voranzubringen.
Quelle: sciencealert
Kommentar hinterlassen