8 Minuten
Zellen sind auf Mitochondrien angewiesen, um Energie zu erzeugen. Neue Forschung verbindet jedoch Fehler bei der mtDNA-Replikation mit einer chronischen Entzündungsreaktion, die mit dem Altern zunimmt. Wissenschaftler des Max-Planck-Instituts für Biologie des Alterns und Kooperationspartner haben menschliche Gewebeproben und altersbedingte Mausmodelle analysiert, um einen molekularen Mechanismus zu identifizieren: Mitochondrien stoßen Fragmente ihrer eigenen DNA in das Zellinnere aus, was dort entzündliche Signalwege aktiviert.
Wissenschaftlicher Hintergrund: mtDNA, Nukleotide und Seneszenz
Mitochondrien besitzen ein eigenes Genom — die mitochondriale DNA (mtDNA) — das während der Mitochondrien-Erneuerung und -Replikation akkurat kopiert werden muss. Für die DNA-Synthese benötigt das Organell die richtigen Monomere. Im Kern der Zelle nutzt die Replikation Deoxyribonukleotide (dNTPs) als Bausteine. Frühere Studien zeigten, dass die Pools dieser dNTPs in gealterten und seneszenten Zellen abnehmen, was die Kopiergenauigkeit beeinträchtigen kann und somit biologische Prozesse stört.
Wenn Deoxyribonukleotide knapp werden, besteht die Gefahr, dass mitochondriale DNA-Polymerasen stattdessen Ribonukleotide (rNTPs) in die wachsende DNA-Kette einbauen. Diese Ribonukleotide verändern die physikalische Stabilität der DNA, weil sie eine andere Zuckerkomponente besitzen; dadurch entstehen lokale Strukturstörungen, einzelne Brüche oder fehlerhafte Kopien. Solche Läsionen kann die Zelle als defekt erkennen. Mitochondrien verfügen über Kontrollmechanismen zur Qualitätsprüfung, und beschädigte mtDNA-Abschnitte werden in manchen Fällen aus dem Organell entfernt und ins Zytosol freigesetzt.
Die Anwesenheit von mtDNA im Zytosol ist nicht neutral: Zytosolische DNA wird von angeborenen Immunrezeptoren erkannt und aktiviert Signalwege, die entzündliche Programme auslösen können. Zu den bekannten Sensoren gehören der cGAS–STING-Weg, der eine Typ-I-Interferon-Antwort induziert, sowie TLR9 in Endosomen, das auf DNA mit CpG-Motiven anspricht. Die Aktivierung dieser Pfade führt zu Zytokinfreisetzung und Rekrutierung von Immunzellen, was langfristig zu Gewebeschäden und fibrotischen Veränderungen beitragen kann.
Aus Sicht der Zellbiologie verbindet dieses Modell metabolische Veränderungen mit Immunreaktionen: Ein Mangel an dNTPs beeinträchtigt die mtDNA-Integrität, dies resultiert in mtDNA-Fragmentierung und -Freisetzung, und schließlich in inflammatorischen Signalen, die das Alterungsbild verschärfen können.
Studiendesign und zentrale Ergebnisse
Die Forschenden kombinierten Analysen an menschlichen Gewebeproben mit genetisch veränderten Mausmodellen, die altersbedingte metabolische Veränderungen nachahmen. Ziel war es nachzuzeichnen, wie ein verändertes Verhältnis von Nukleotiden die mtDNA-Integrität beeinflusst und über welche Mechanismen dies zu Entzündung führt. Mittels biochemischer Assays, hochauflösender Sequenzierung, histologischer Färbungen und Zellbiologie-Arbeiten konnten sie mehrere konsistente Beobachtungen machen.
In Geweben älterer Mäuse und in Versuchsbedingungen mit künstlich reduzierten dNTP-Pools fand sich eine deutlich höhere Rate an in die mtDNA eingebetteten Ribonukleotiden. Solche fehlerhaften Einbauten führten zu erhöhten Bruchraten, zu ungewöhnlichen Konformationen des mtDNA-Kreises und zu vermehrter Fragmentierung. Die so entstandenen, unvollständigen mtDNA-Moleküle wiesen eine größere Neigung zur Ausschleusung aus dem Mitochondrium auf; dieser Vorgang wurde in Zellbildgebungs-Experimenten und durch die Quantifizierung von zytosolischer DNA nachgewiesen.
Die Forschenden nutzten zusätzlich genetische Reporter und Immunmarkierungen, um Signalwege nachzuverfolgen: Freigesetzte mtDNA aktivierte klassische DNA-Sensoren, erhöhte Interferon- und proinflammatorische Zytokinspiegel und förderte die Expression von Genen, die mit Immunantwort und Fibrose assoziiert sind. Morphologisch zeigten betroffene Nieren Gewebeverhärtung und Narbenbildung, typische Merkmale chronisch entzündlicher Schädigung.
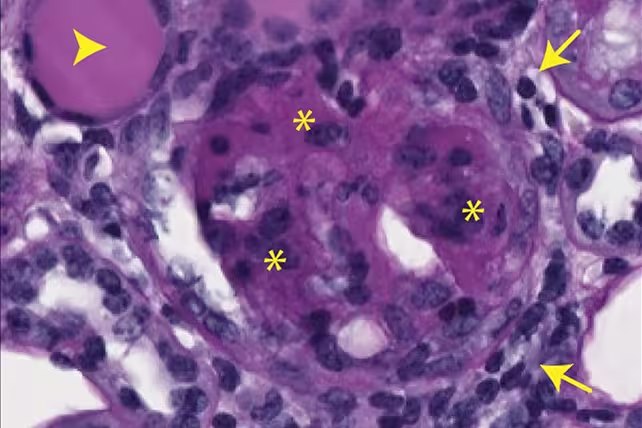
Nachweis von Nierenvernarbung bei Mäusen, verursacht durch freigesetzte mtDNA. (Max Planck Institute for Biology of Ageing)
Zusammengefasst verbinden die Ergebnisse einen metabolischen Engpass — sinkende Verfügbarkeit von dNTPs — mit einer molekularen Abfolge: fehlerhafte Replikation der mtDNA, Fragmentierung und Ejektion von mtDNA in das Zytosol sowie anschließende Aktivierung angeborener Immunantworten. Die Arbeit liefert damit eine plausible molekulare Brücke zwischen mitochondrialer Dysfunktion und entzündlichen Phänotypen im Alter.
Wichtige Aussagen der Forschenden
Thomas Langer vom Max-Planck-Institut für Biologie des Alterns fasst die Bedeutung so zusammen: Stoffwechselstörungen, die Nukleotid-Pools verändern, können eine Kaskade auslösen, die in seneszenten Zellen und gealterten Organen Entzündungen fördert. Dies eröffnet potenzielle molekulare Angriffsflächen für Interventionen. Dusanka Milenkovic, ebenfalls am Institut, weist darauf hin, dass Therapien, die bereits bei bestimmten mitochondrialen Erkrankungen angewendet werden — etwa die Gabe von DNA-Vorläufern oder Nukleosiden — geprüft werden sollten, ob sie auch altersbedingte Entzündungsprozesse reduzieren können.
Beide Forscher betonen jedoch die Notwendigkeit vorsichtiger Validierung: Die Verabreichung von Nukleosiden kann in unterschiedlichen Kontexten unterschiedliche Effekte haben, und die langfristigen Folgen einer Manipulation von dNTP-Pools müssen in präklinischen Modellen gründlich untersucht werden, bevor klinische Studien gestartet werden. Ebenso ist es wichtig, Mechanismen für die Entfernung oder Reparatur eingebauter Ribonukleotide, beispielsweise durch RNase-H-Mechanismen oder spezialisierte Reparaturpfade, weiter zu erforschen.
Bedeutung für Gesundheit und weitere Forschung
Der beschriebene Mechanismus bietet eine nachvollziehbare Erklärung dafür, wie sich über Jahrzehnte eine low-grade, also niedriggradige, chronische Entzündung aufbauen kann — ein Phänomen, das als Inflammaging bezeichnet wird und zur Entstehung häufiger altersassoziierter Erkrankungen beiträgt. Beispiele hierfür sind bestimmte Krebsarten, Herz-Kreislauf-Erkrankungen und neurodegenerative Leiden wie Alzheimer oder Parkinson. Mitochondriale Fehlfunktionen und persistierende Immunaktivierung scheinen in vielen dieser Krankheitsbilder eine Rolle zu spielen.
Therapeutisch betrachtet lassen sich daraus mehrere Ansatzpunkte ableiten: Erstens könnte man versuchen, die Einbaurate von Ribonukleotiden in die mtDNA zu reduzieren, zum Beispiel durch Stabilisierung der dNTP-Pools. Zweitens wäre es denkbar, die Reparatur- und Qualitätskontrollmechanismen der Mitochondrien zu stärken, sodass beschädigte mtDNA fragmente gar nicht erst in das Zytosol gelangen. Drittens können direkte Modulatoren der DNA-Erkennungswege oder Anti-entzündungsstrategien die nachgeschaltete Antwort dämpfen und so Gewebeschaden verhindern.
Konkrete experimentelle Richtungen umfassen dabei die Evaluierung von Nukleosid- oder Deoxynukleotid-Supplementen, die Modulation von Enzymen wie der Ribonukleotid-Reduktase (RNR) zur Erhöhung der dNTP-Synthese, sowie Eingriffe in Proteine, die für mtDNA-Reparatur und -Erhaltung zuständig sind (beispielsweise die mitochondriale DNA-Polymerase POLG und Reparatursysteme wie RNase H). Diese Ansätze müssen jedoch nach Sicherheitsprofilen bewertet werden: Eine Überversorgung mit Nukleotiden kann unerwünschte Proliferationseffekte begünstigen oder andere Stoffwechselwege stören.
Weitere zentrale Fragestellungen für die Forschung sind:
- Wie häufig operiert dieser Weg während des physiologischen Alterns beim Menschen gegenüber in spezifischen Krankheiten?
- Welche Zelltypen sind besonders anfällig für dNTP-Verluste und mtDNA-Freisetzung — sind es hochmetabolische Zellen wie Neurone, Muskelzellen oder Nierepithelzellen?
- Welche genetischen oder umweltbedingten Faktoren verstärken dieses Phänomen, und inwieweit besteht Variabilität zwischen Individuen?
Die Beantwortung dieser Fragen erfordert groß angelegte Studien mit menschlichen Proben, longitudinale Kohortenanalysen und zusätzliche Tiermodelle, die unterschiedliche Aspekte der Nukleotid- und Mitochondrienbiologie abbilden. Ebenfalls wichtig sind translational ausgerichtete Versuche, die Wirksamkeit und Sicherheit möglicher Interventionen prüfen.
Methodische und konzeptionelle Grenzen
Obwohl die Daten überzeugend sind, bestehen Einschränkungen, die beachtet werden sollten. Tiermodelle reproduzieren nicht immer vollständig die Komplexität des menschlichen Alterns. Zudem ist die Kausalitätskette — von dNTP-Mangel über rNTP-Einbau bis zur klinisch relevanten Entzündung — in biologischen Systemen vielschichtig und von weiteren Faktoren abhängig, wie zellulären Reparaturmechanismen, Autophagie-Aktivität oder dem Zustand des Immunsystems.
Technische Limitationen betreffen die Quantifizierung und Lokalisierung freigesetzter mtDNA-Fragmente: Es bleibt schwierig, genau zu bestimmen, welche molekularen Formen (lineare Fragmente, zirkuläre Bruchstücke, mtDNA assoziiert mit Membransäumen) am stärksten immunstimulierend sind. Ferner können andere Quellen für zytosolische DNA — etwa genomische DNA bei genetischer Instabilität — zu ähnlichen Signalen führen und müssen klar unterschieden werden.
Schließlich ist die therapeutische Übersetzung komplex: Eine Maßnahme, die in einem Organsystem vorteilhaft wirkt, kann in einem anderen Schaden anrichten. Deshalb ist ein differenziertes Verständnis der Dosis-Wirkungs-Beziehung ebenso notwendig wie die Identifikation von Biomarkern, die anzeigen, welche Patientengruppen von einer Intervention am meisten profitieren würden.
Fazit
Die Studie verknüpft einen metabolischen Engpass — die Abnahme von Deoxyribonukleotiden — mit einem klaren molekularen Pfad zur Entzündung über mtDNA-Instabilität und deren Ausschleusung. Diese Einsicht öffnet vielversprechende Möglichkeiten, gezielt die Integrität des mitochondrialen Genoms zu erhalten und so potentiell die entzündungsbedingte Funktionsverschlechterung im Alter zu verlangsamen. Translationale Schritte sind notwendig, um zu prüfen, ob Nukleotid-Supplementation oder andere Interventionen in präklinischen und klinischen Studien tatsächlich messbare gesundheitliche Vorteile für ältere Menschen bringen.
Langfristig könnte das Verständnis dieses Mechanismus zur Entwicklung von Biomarkern führen, die frühzeitig eine mitochondriale Dysfunktion oder eine gefährliche Neigung zu mtDNA-Freisetzung anzeigen. Solche Marker würden ermöglichen, präventive Therapien gezielt bei Menschen einzusetzen, die am meisten davon profitieren, und so das Risiko altersassoziierter entzündlicher Erkrankungen zu senken.
Quelle: sciencealert
Kommentar hinterlassen